22 时间序列分析
22.1 什么是时间序列分析?
在研究基因表达谱或者蛋白表达谱时,经常会涉及到对时间序列的分析。例如,不同的基因或蛋白表达水平随时间表现出怎样的动力学特征,怎样挖掘潜在的时间特征?
本篇让我们来看一个能够分析基因表达谱或者蛋白表达谱的时间动力学特征的R包-Mfuzz。它能够识别表达谱的潜在时间序列模式,并将相似模式的基因聚类,以帮助我们了解基因的动态模式和它们功能的联系
对于微阵列数据的分析,经常使用聚类技术。大多数此类方法都基于数据的硬聚类,其中一个基因(或样本)被分配到一个聚类中。然而,硬聚类存在一些缺点,例如对噪声敏感和信息丢失。相反,软聚类方法可以将一个基因分配给几个聚类。它们可以克服传统硬聚类技术的缺点并提供更多优势。Mfuzz,使用模糊 c 均值算法实现了软聚类
本文我们就来讨论一下时间序列分析是如何进行的以及如何对其进行解读。
22.2 绘图前的数据准备
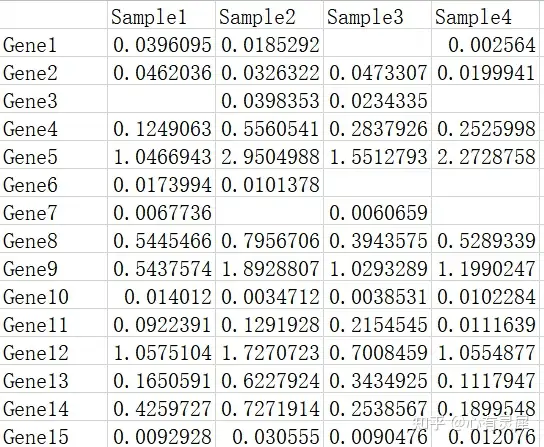
demo数据可以在https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/mfuzz/all_gene_fpkm.xls下载。
数据包含2个维度,数据通常来源于搜库结果定量表。每一行代表一个基因,每一列代表一个分组。
22.3 R语言怎么做时间序列分析
# Mfuzz包的安装方式为:BiocManager::install("Mfuzz")
library(Mfuzz)
# 读取时间序列分析数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/mfuzz/all_gene_fpkm.xls",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
row.names = 1
)
# 构建对象,填补缺失值,标准化等
dm <- data.matrix(df) # 数据框转换为矩阵
ESet <- new("ExpressionSet",exprs = dm) # 构建对象
ESet <- filter.NA(ESet, thres=0.25) # 过滤缺失值超过“25%”的基因
ESet <- fill.NA(ESet,mode="knn") # knn算法填补缺失值
ESet <- filter.std(ESet,min.std=0) # 根据标准差去除样本间差异太小的基因
gene.s <- standardise(ESet) # 标准化
exprs(gene.s) # 查看处理后的数据
# 聚类
c <- 6 # 设置聚类个数
m <- mestimate(gene.s) # 评估出最佳的m值
set.seed(123) # 设置随机种子,防止每次聚类的结果都不一样,无法复现
cl <- mfuzz(gene.s, c = c, m = m)
cl # 查看每个基因聚到哪个类当中
cl$size # 查看每个cluster中的基因个数
cl$membership # 查看基因和cluster之间的membership。如果两个基因对于一个特定的cluster都有高的membership score,那么他们通常来说表达模式是相似的
# 绘图
mfuzz.plot(
gene.s,
cl,
mfrow=c(2,3),# 图形排列方式,2行3列
new.window= FALSE) # 是否在新窗口打开22.4 BioLadder生信云平台在线做时间序列分析
不想写代码?可以用BioLadder生信云平台在线做时间序列分析。
网址:
时间序列分析-BioLadder生物信息在线分析可视化云平台www.bioladder.cn/web/#/chart/8
22.5 时间序列分析结果解读
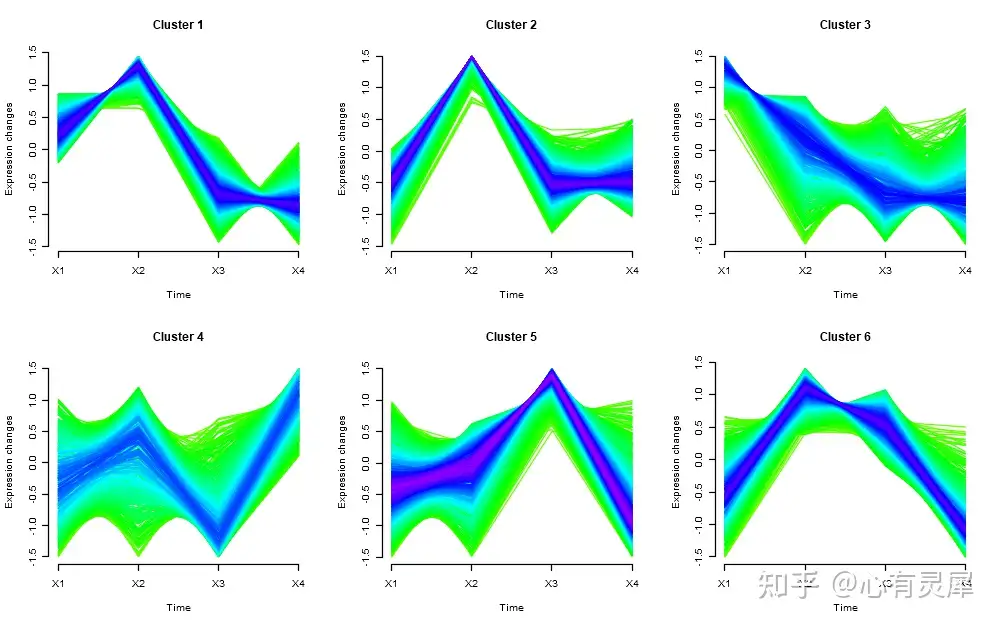
如上过程基于基因表达值进行了聚类,对于每个簇中的基因,具有相似的时间表达特征。随后,即可从图中识别一些重要的聚类簇,比方说簇中基因随时间表达趋势增加或减少,以及在特定时间出现了更高或更低的表达等,以建立和观察的表型的联系。